
 bei Colitis ulcerosa und Morbus Crohn")
Colitis ulcerosa (UC) und Morbus Crohn (CD) sind chronisch-entzündliche Darmerkrankungen (IBD), die zu fortschreitenden Darmschäden und massiven Einschränkungen im täglichen Leben der Patient:innen führen können [2, 3]. Mit der neuen Doppelzulassung von RINVOQ®(Upadacitinib) steht Patient:innen in der Schweiz erstmals ein oraler, selektiver Januskinase1 (JAK1)-Inhibitor zur Behandlung der UC und des CD zur Verfügung [1]. Die Resultate der klinischen Zulassungsstudien zeigen ein schnelles Ansprechen innert Tagen, anhaltende Remission über 52 Wochen und lassen langfristige stringente Therapieziele wie die Mukosaheilung bei beiden Indikationen möglich werden [1, 4-8].
Seit dem 7. Juni 2024 ist RINVOQ® zur Behandlung von mittelschwerer bis schwerer aktiver UC sowie mittelschwerem bis schwerem aktivem CD zugelassen – dies bei erwachsenen Patient:innen, die auf mindestens ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder dieses nicht vertragen haben, oder bei denen eine solche Therapie kontraindiziert ist [1]. Die Zulassung stützt sich auf die Ergebnisse von mehreren globalen Phase-3-Studien und zeigt zusammenfassend, dass RINVOQ® als 8-wöchige (bei UC), respektive 12-wöchige (bei CD), Induktionstherapie mit einer darauffolgenden Erhaltungstherapie bei Patient:innen in beiden Indikationen gut verträglich ist und die primären wie auch sekundären Endpunkte erreicht wurden [4, 5, 7]. Im Folgenden werden Daten zu den Patient:innenpopulationen vorgestellt, welche vor der RINVOQ®-Therapie ungenügend auf die Behandlung mit einem Biologikum angesprochen haben (bio-IR), d.h. unzureichendes Ansprechen, Verlust des Ansprechens oder Unverträglichkeit gegenüber ≥1 Biologikum [5, 7].
Schnelles Ansprechen, dauerhafte Remission und Mukosaheilung bei UC
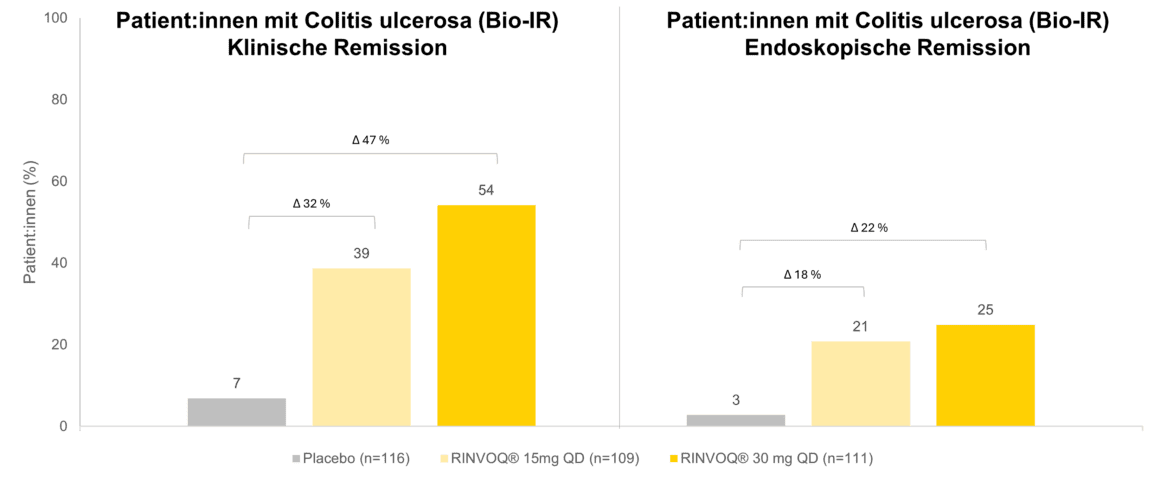
Die Induktionsstudien zu UC U-ACHIEVE und U-ACCOMPLISH schlossen insgesamt 988 Patient:innen ein (RINVOQ® n=660, Placebo n=328), davon waren rund die Hälfte bio-IR (51,5 % RINVOQ®, 50,9 % Placebo) [4, 6]. Bereits nach dem ersten Tag zeigte sich eine signifikante Symptomverbesserung (Stuhlgangshäufigkeit, rektale Blutungen) mit RINVOQ® (45 mg/Tag) im Vergleich zu Placebo† [6]. In der Erhaltungsstudie U-ACHIEVE wurden 681 Patient:innen mit Ansprechen in der Induktionsphase weitergeführt mit 15 mg/Tag bzw. 30 mg/Tag RINVOQ® (n=225 bzw. n= 233; Placebo n=223; davon 48 % bio-IR unter RINVOQ® und 52 % unter Placebo) [5]. In Woche 52 erreichten 38,7 % (15 mg) bzw. 54,2 % (30 mg) der mit RINVOQ® behandelten bio-IR-Patient:innen eine klinische Remission#, gegenüber 6,9 % unter Placebo (Abbildung. 1). Eine endoskopische Verbesserung£ wurde sogar bei 43,9 % (15 mg) bzw. 59,2 % (30 mg) beobachtet, gegenüber 7,4 % unter Placebo [5]. Des Weiteren konnten 61 % (15 mg) bzw. 73 % (30 mg) der Patient:innen, welche sich nach der Induktionstherapie in klinischer Remission befanden, die klinische Remission bis zu Woche 52 dauerhaft erhalten [5].
Schnelles Ansprechen, anhaltende Remission und Mukosaheilung auch bei CD
Die CD-Induktionsstudien U-EXCEL und U-EXCEED schlossen 1021 Patient:innen ein (RINVOQ® n=350, Placebo n=176 bzw. RINVOQ® n=324, Placebo n=171), davon 46,0 % bio-IR unter RINVOQ® und 44,3 % unter Placebo, bzw. 100 % bio-IR [7]. Am Ende der 12-wöchigen Induktionsphase mit 45 mg/Tag RINVOQ® erreichten 47 % (U-EXCEL) bzw. 40 % (U-EXCEED) der bio-IR-Patient:innen eine klinische Remission§, gegenüber 14 % unter Placebo in beiden Studien [1]. Die angeschlossene Erhaltungsstudie U-ENDURE wurde mit 502 Patient:innen weitergeführt, die während der Induktionsphase angesprochen hatten [7]. Diese erhielten 15 mg/Tag bzw. 30 mg/Tag RINVOQ® (n=169 bzw. n=168) oder Placebo (n=165), davon 74,5 % bio-IR unter RINVOQ® und 76,4 % unter Placebo [7]. In Woche 52 hatten 32,3 % (15 mg) bzw. 42,5 % (30 mg) der bio-IR-Patient:innen eine klinische Remission§ erreicht, im Gegensatz zu 8,7 % unter Placebo [7]. Dabei konnten 44 % (15 mg) bzw. 60 % (30 mg) der Patient:innen, welche sich nach der Induktionstherapie in klinischer Remission befanden, die klinische Remission bis zu Woche 52 erhalten [9]. Eine Mukosaheilung, definiert als ulkusfreie Endoskopie, wurde bei 12 % (15 mg) bzw. 20 % (30 mg) der RINVOQ® behandelten bio-IR-Patient:innen und 2 % der bio-IR-Patient:innen unter Placebo beobachtet [1].
Das Sicherheitsprofil von RINVOQ® zeigt ein positives Nutzen-Risiko-Verhältnis
RINVOQ® war in den placebokontrollierten klinischen UC- und CD-Studien gut verträglich und es wurden keine neuen Sicherheitsrisiken beobachtet verglichen zu den anderen zugelassenen Indikationen in der Rheumatologie und Dermatologie [1, 5, 7]. Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (≥3 % der Patient:innen mit UC oder CD) unter RINVOQ®(45 mg, 30 mg oder 15 mg) waren Infektionen der oberen Atemwege, Fieber, Erhöhung der Kreatinphosphokinase, Anämie, Kopfschmerzen, Akne, Herpes Zoster, Neutropenie, Ausschlag, Pneumonie, Hypercholesterinämie, Bronchitis, erhöhte Aspartattransaminase, Ermüdung, Follikulitis, erhöhte Alanintransaminase, Herpes Simplex und Grippe [1]. Die Wirkung und Sicherheit von RINVOQ®wird weiterhin in laufenden Langzeitstudien untersucht [5, 7]. Angesichts des erhöhten Risikos für schwere Infektionen, Myokardinfarkt und maligne Erkrankungen im Zusammenhang mit JAK-Inhibitoren bei Patient:innen über 65 Jahren sollte RINVOQ® bei UC- und CD-Patient:innen in dieser Altersgruppe oder mit kardiovaskulären Risikofaktoren (inkl. gegenwärtige Raucher:innen oder Ex-Raucher:innen) oder Risikofaktoren für maligne Erkrankungen mit besonderer Vorsicht angewendet werden [1]. Zusammenfassend zeigen die Daten der UC- und CD-Zulassungsstudien, dass beide RINVOQ®-Erhaltungsdosen signifikant wirksamer waren als Placebo – dies in Bezug auf wichtige klinische, endoskopische und histologische Ergebnisse [5, 7]. Gleichzeitig wurde ein gut untersuchtes Sicherheitsprofil ohne neue Sicherheitsrisiken in den gesamten Studienpopulationen über 52 Wochen aufrechterhalten [5, 7].
Fazit
Für UC- und CD-Patient:innen bietet RINVOQ® nach Versagen von mindestens einem Biologikum eine vielversprechende neue Behandlungsoption mit schnellem Wirkeintritt, anhaltender Remission und Mukosaheilung über mindestens 52 Wochen. Die Doppelzulassung von RINVOQ® für UC und CD ist damit ein weiterer Meilenstein, welcher das Erreichen der kurz-, mittel- und auch der langfristigen Therapieziele einschliesslich endoskopischer Heilung, wie diese im Treat-to-Target Algorithmus von STRIDE II empfohlen werden, ermöglichen kann [4-8, 10].
† Diese Analyse enthält Daten von Patient:innen, die zuvor noch nicht auf ein Biologikum inadäquat angesprochen hatten und dementsprechend nach Schweizer Zulassung off-label behandelt wurden.
# UC klinische Remission = Adapted Mayo Score (Adaptierter Mayo-Score ≤2, mit SFS ≤1 und nicht grösser als der Ausgangswert, RBS=0 und ESS ≤1 ohne Friabilität. ESS, Endoskopischer Score; RBS, rektaler Blutungs-Score (rectal bleeding score); SFS, Stuhlfrequenz-Score (stool frequency score)) [4]
£ UC endoskopische Verbesserung = ESS ≤ 1 ohne Friabilität. ESS, Endoskopischer Score [5].
§ CD klinische Remission= SF/APS (durchschnittliche tägliche SF ≤ 2,8 UND durchschnittliche tägliche APS ≤ 1, mit beiden Werten nicht schlechter als der Ausgangswert. APS, abdominaler Schmerz-Score (abdominal pain score); SF, Stuhlfrequenz) [7]
Literatur:
1. Aktuelle Fachinformation von RINVOQ® (Upadacitinib) auf www.swissmedicinfo.ch.
2. Dolinger, M., J. Torres, and S. Vermeire, Crohn’s disease. Lancet, 2024. 403(10432): p. 1177-1191.
3. Le Berre, C., S. Honap, and L. Peyrin-Biroulet, Ulcerative colitis. Lancet, 2023. 402(10401): p. 571-584.
4. Danese, S., et al., Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet, 2022. 399(10341): p. 2113-2128. Incl. Suppl.
5. Vermeire, S., et al., Efficacy and safety of upadacitinib maintenance therapy for moderately to severely active ulcerative colitis in patients responding to 8 week induction therapy (U-ACHIEVE Maintenance): overall results from the randomised, placebo-controlled, double-blind, phase 3 maintenance study. Lancet Gastroenterol Hepatol, 2023. 8(11): p. 976-989. Incl. Suppl.
6. Loftus, E.V., Jr., et al., Upadacitinib Therapy Reduces Ulcerative Colitis Symptoms as Early as Day 1 of Induction Treatment. Clin Gastroenterol Hepatol, 2023. 21(9): p. 2347-2358 e6.
7. Loftus, E.V., Jr., et al., Upadacitinib Induction and Maintenance Therapy for Crohn’s Disease. N Engl J Med, 2023. 388(21): p. 1966-1980. Incl. Suppl.
8. Colombel, J.F., et al., Upadacitinib Reduces Crohn’s Disease Symptoms Within the First Week of Induction Therapy. Clin Gastroenterol Hepatol, 2024.
9. Schreiber SW. et al. P630. Upadacitinib Improves Clinical Outcomes in Patients with Moderate to Severely Active Crohn’s Disease Irrespective of Previous Failure to Respond to Biologics or Conventional Therapies. Poster Presented at ECCO March 2023. Journal of Crohn’s and Colitis. 2023(17)S1:i759–i760.
10. Turner, D., et al., STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology, 2021. 160(5): p. 1570-1583.
Die Referenzen können durch Fachpersonen bei medinfo.ch@abbvie.com angefordert werden.
Dieser Beitrag entstand mit finanzieller Unterstützung der AbbVie AG, Alte Steinhauserstrasse 14, Cham.
CH-RNQG-240117 08/2024
Kurzfassung Fachinformation RINVOQ® (Upadacitinib)
I: Erwachsene mit mittelschwerer bis schwerer aktiver Rheumatoider Arthritis (RA) mit unzureichendem Ansprechen oder Unverträglichkeit auf ein oder mehrere konventionelle synthetische krankheitsmodifizierende Antirheumatika (csDMARDs). In Kombination mit Methotrexat oder anderen csDMARDs oder als Monotherapie; Erwachsene mit aktiver Psoriasis-Arthritis (PsA) mit unzureichendem Ansprechen oder Unverträglichkeit auf ein oder mehrere Antirheumatika (DMARDs). Als Monotherapie oder in Kombination mit nicht biologischen DMARDs; Erwachsene mit aktiver Ankylosierender Spondylitis (AS, Morbus Bechterew) mit unzureichendem Ansprechen auf nicht-steroidale Antirheumatika (NSAIDs); Erwachsene mit mittelschwerer bis schwerer atopischer Dermatitis (AD), wenn eine Therapie mit konventionellen topischen Medikamenten keine angemessene Krankheitskontrolle ermöglicht oder nicht angewendet werden kann; Erwachsene mit mittelschwerer bis schwerer aktiver Colitis ulcerosa (UC) die auf mindestens ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder dieses nicht vertragen oder bei denen eine solche Therapie kontraindiziert ist; Erwachsene mit mittelschwerem bis schwerem aktivem Morbus Crohn (CD) die auf mindestens ein Biologikum unzureichend angesprochen haben, nicht mehr darauf ansprechen oder dieses nicht vertragen oder bei denen eine solche Therapie kontraindiziert ist. D: Für RA-, PsA-, AS- und AD-Patienten: Empfohlene orale Dosis 15 mg einmal täglich; Für UC- und CD-Patienten: Induktionsdosis beträgt 45 mg einmal täglich für 8 Wochen bei UC und 12 Wochen bei CD. Bei unzureichendem Ansprechen nach der Induktionsphase kann die Induktion um 8 Wochen mit 45 mg täglich bei UC oder um 12 Wochen mit 30 mg täglich bei CD verlängert werden. Erhaltungsdosis beträgt 15 mg oder 30 mg einmal täglich. Bei Patienten mit hoher Krankheitsaktivität, verlängerter Induktionsphase oder welche unzureichend auf 15 mg ansprechen, kann eine Dosis von 30 mg in Betracht gezogen werden. Bei einer längeren Induktionsphase und bei der Wahl der Erhaltungsdosis muss das Risiko für MACE, VTE und maligne Erkrankungen berücksichtigt werden. Bei Patienten, die starke CYP3A-Hemmer erhalten, mit leichter oder mittelschwerer Leberinsuffizienz (Child-Pugh A oder B) oder schwerer Niereninsuffizienz: Induktionsdosis von 30 mg einmal täglich und Erhaltungsdosis von 15 mg einmal täglich. Für Patienten ≥ 65 Jahre: Erhaltungsdosis von 15 mg einmal täglich; Anwendung bei absoluter Lymphozytenzahl (ALC) <500 Zellen/mm3, absoluter Neutrophilenzahl (ANC) <1000 Zellen/mm3, Hämoglobinspiegel (Hb) <8 g/dl, schwerer Leberinsuffizienz (Child-Pugh C) oder terminaler Niereninsuffizienz nicht empfohlen; Unterbruch bei schwerwiegender Infektion. KI: Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe. Aktive Tuberkulose. W: Bei folgenden RA-, PsA-, AS- und AD-Patienten nur anwenden, wenn keine geeigneten Behandlungsalternativen zur Verfügung stehen: Über 65 Jahre, (ehemalige) Raucher, Risikofaktoren für maligne Erkrankungen (einschliesslich Nicht-melanozytärer Hautkrebs, NMSC) oder mit kardiovaskulären Risikofaktoren; Bei folgenden UC- und CD-Patienten nur mit besonderer Vorsicht anwenden: Über 65 Jahre, (ehemalige) Raucher, Risikofaktoren für maligne Erkrankungen (einschliesslich Nicht-melanozytärer Hautkrebs, NMSC) oder mit kardiovaskulären Risikofaktoren; Im Vergleich zur 15-mg-Dosis ist bei der Behandlung mit einer Dosis von 30 mg einmal täglich mit einem erhöhten Risiko von Nebenwirkungen, einer höheren Rate schwerwiegender Infektionen und einer höheren Rate von malignen Erkrankungen zu rechnen; Anwendung bei Patienten mit einer aktiven, schweren Infektion vermeiden. Auf Anzeichen und Symptome achten. Behandlung unterbrechen, bis die Infektion unter Kontrolle ist. Vor Beginn der Therapie ein Screening auf Tuberkulose (TB) und virale Hepatitis durchführen und auf eine Reaktivierung überwachen. Bei Patienten mit zuvor unbehandelter latenter TB muss vor der Behandlung eine TB-Prophylaxe eingeleitet werden. Bei Entwicklung eines Herpes zoster Abbrechen der Therapie bis zum Abklingen der Episode in Erwägung zu ziehen. Aktualisierung des Impfstatus, einschliesslich Impfungen gegen Varizellen/Zosterinfektionen, vor Beginn der Therapie empfohlen. Anwendung attenuierter Lebendimpfstoffe während oder unmittelbar vor der Therapie nicht empfohlen; Patienten mit Anzeichen und Symptomen thromboembolischer Ereignisse untersuchen und Behandlung bei Verdacht abbrechen; Behandlung abbrechen, wenn klinisch signifikante Überempfindlichkeitsreaktion auftritt; Anwendung mit Vorsicht bei Patienten mit Risiko auf eine gastrointestinale Perforation; Überwachung von ANC, ALC, Hb, Lipidparametern und Lebertransaminasen; Arzneimittelrückstände im Stuhl oder in den Ausscheidungen des Stoma möglich. IA: Vorsicht bei gleichzeitiger Verabreichung mit starken CYP3A4 Inhibitoren. Gleichzeitige Verabreichung mit starken CYP3A4 Induktoren nicht empfohlen (z.B. Rifampicin). SS: Während Schwangerschaft oder Stillzeit nicht anwenden. UW: Sehr Häufig (≥1/10): Infektionen der oberen Atemwege (bei AD), Akne (bei AD). P: 28 Retardtabletten à 15 mg, 30 mg und 45 mg. Liste B: Kassenzulässig. Z: AbbVie AG, Alte Steinhauserstrasse 14, 6330 Cham, Tel. (+41) 41 399 15 00. (V7) Ausführliche Informationen, siehe Arzneimittel-Fachinformation: www.swissmedicinfo.ch


Comments are closed.