Beschreibung
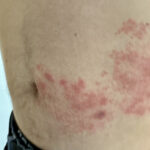
Koagulopathien, Purpura und andere hämorrhagische Störungen bilden eine Gruppe von hämatologischen Erkrankungen, die durch eine abnorme Blutungsneigung und gestörte Blutgerinnungsmechanismen gekennzeichnet sind. Diese Störungen äußern sich in einem Spektrum von Symptomen, die je nach Erkrankung variieren können. Zu den häufigen Symptomen gehören unerklärliche und übermäßige Blutungen, leichte Blutergüsse, Petechien (kleine rote oder violette Flecken auf der Haut) und anhaltende Blutungen bei kleineren Verletzungen. Innerhalb dieser Kategorie gibt es verschiedene Typen, die jeweils durch unterschiedliche zugrunde liegende Mechanismen gekennzeichnet sind.
Die Prävalenz dieser Störungen variiert weltweit, wobei einige relativ selten sind, während andere in der klinischen Praxis häufiger anzutreffen sind. Historisch gesehen haben sich das Verständnis und die Kategorisierung dieser Störungen erheblich weiterentwickelt. Frühe Beobachtungen hämorrhagischer Tendenzen reichen Jahrhunderte zurück, und die Fortschritte in der medizinischen Wissenschaft haben tiefere Einblicke in ihre Pathophysiologie und Behandlung ermöglicht.
Die Komplikationen, die sich aus diesen Störungen ergeben, können schwerwiegend und vielfältig sein. Chronischer Blutverlust kann zu Blutarmut (Anämie) führen, die sich in Müdigkeit, Schwäche und Blässe äußert. Außerdem können unbehandelte oder schlecht behandelte hämorrhagische Störungen zu lebensbedrohlichen Blutungen, Organschäden oder neurologischen Beeinträchtigungen führen.
Eine genaue Diagnose dieser Erkrankungen ist für eine wirksame Behandlung von größter Bedeutung. Die klinische Beurteilung, die Erhebung der Krankengeschichte und spezielle Labortests sind von entscheidender Bedeutung für die Bestimmung der spezifischen Störung und ihres Schweregrads. Die Behandlungsstrategien sind auf die jeweilige Diagnose zugeschnitten und können unter anderem Ersatztherapien, die Verabreichung von Gerinnungsfaktoren, Antifibrinolytika und Thrombozytentransfusionen umfassen.
Die Ursachen für Koagulopathien, Purpura und andere hämorrhagische Störungen sind vielfältig und umfassen genetische Mutationen, Autoimmunreaktionen, medikamentenbedingte Faktoren und Grunderkrankungen. Zu den Risikofaktoren gehören eine familiäre Vorbelastung mit Blutungsstörungen, genetische Veranlagungen, die Einnahme gerinnungshemmender Medikamente und bestimmte chronische Krankheiten.
Die Vorbeugung dieser Erkrankungen umfasst häufig Maßnahmen wie genetische Beratung bei Personen mit familiärer Vorbelastung, regelmäßige ärztliche Untersuchungen zur Früherkennung und eine sorgfältige Behandlung der Grunderkrankungen. Praktiken der Arzneimittelsicherheit und der Patientenaufklärung sind für die Vorbeugung von arzneimittelbedingten Hämorrhagieerkrankungen von entscheidender Bedeutung.
Die Biologie dahinter
Um einen Einblick in die Pathophysiologie dieser Störungen zu erhalten, ist es unerlässlich, sich mit den komplizierten Mechanismen der Hämostase, dem körpereigenen System zur Blutungsregulierung, zu befassen. Die Hämostase hängt von einem empfindlichen Gleichgewicht zwischen gerinnungsfördernden und gerinnungshemmenden Faktoren ab, um die Blutzirkulation aufrechtzuerhalten und gleichzeitig eine übermäßige Blutung zu verhindern.
Zu den Schlüsselkomponenten der Hämostase gehören Blutplättchen, Gerinnungsfaktoren und das Gefäßendothel. Thrombozyten sind winzige Zellfragmente, die an der Stelle einer Gefäßverletzung zusammenkommen und einen provisorischen Pfropfen bilden. Die Gerinnungskaskade orchestriert eine Abfolge von enzymatischen Reaktionen, an denen verschiedene Gerinnungsfaktoren beteiligt sind und die schließlich in der Bildung eines stabilen Fibringerinnsels gipfeln. Das vaskuläre Endothel, das die Blutgefäße auskleidet, spielt eine zentrale Rolle bei der Aufrechterhaltung der Gefäßintegrität und der Steuerung der Gerinnung.
Störungen in diesem sorgfältig ausbalancierten System können Koagulopathien, Purpura und hämorrhagische Diathesen auslösen. Bei Koagulopathien kommt es häufig zu Anomalien in der Produktion oder Funktion von Gerinnungsfaktoren, die entweder zu einer abnormen Gerinnselbildung (Thrombose) oder zu einer Blutungsneigung führen. Purpura, typischerweise eine purpurne oder rötliche Verfärbung der Haut aufgrund von subkutanen oder Schleimhautblutungen, kann verschiedene Ursachen haben, darunter auch eine Fehlfunktion oder Insuffizienz der Blutplättchen. Hämorrhagische Diathesen umfassen ein Spektrum von Blutungsstörungen, die häufig durch multifaktorielle Störungen des Gerinnungsprozesses gekennzeichnet sind.
Arten und Symptome
Innerhalb der Koagulopathien, der Purpura und der hämorrhagischen Störungen gibt es verschiedene Typen, die jeweils durch spezifische Symptome und klinische Präsentationen gekennzeichnet sind. Das Verständnis dieser verschiedenen Erscheinungsformen ist für eine genaue Diagnose und Behandlung von entscheidender Bedeutung.
Koagulopathien:
Hämophilie: Die Hämophilie ist eine anerkannte Gerinnungsstörung, von der vor allem Männer betroffen sind. Sie kann in Hämophilie A (Faktor-VIII-Mangel) und Hämophilie B (Faktor-IX-Mangel) eingeteilt werden. Zu den Symptomen gehören wiederkehrende Gelenk- und Muskelblutungen, übermäßige Blutergüsse und verlängerte Blutungen nach Verletzungen oder Operationen. Eine häufige Komplikation ist die hämophile Arthropathie, die durch Gelenkdeformitäten und chronische Schmerzen gekennzeichnet ist.
Von-Willebrand-Krankheit (VWD): Die VWD ist eine weitere vererbte Koagulopathie, die in der Regel milder verläuft als die Hämophilie. Sie entsteht durch einen Mangel oder eine Störung des von-Willebrand-Faktors (VWF), der eine entscheidende Rolle bei der Adhäsion von Blutplättchen spielt. Zu den Symptomen gehören leichte Blutergüsse, Nasenbluten, starke Menstruationsblutungen bei Frauen und in schweren Fällen spontane Blutungen der Schleimhäute.
Purpura:
Idiopathische thrombozytopenische Purpura (ITP): Die ITP ist durch eine Abnahme der Blutplättchenzahl gekennzeichnet, die zu Petechien (kleine, rote oder violette Punkte auf der Haut), Purpura und Schleimhautblutungen führt. Sie tritt häufig nach Virusinfektionen und Autoimmunreaktionen auf.
Thrombotische thrombozytopenische Purpura (TTP): TTP ist eine seltene, aber lebensbedrohliche Erkrankung, bei der es zu einer Thrombozytenaggregation in kleinen Blutgefäßen kommt. Bei den Patienten können Purpura, Petechien, Fieber, neurologische Symptome und Nierenfunktionsstörungen auftreten.
Hämorrhagische Diathese:
Disseminierte intravasale Gerinnung (DIC): Bei der DIC handelt es sich um ein komplexes Syndrom, bei dem eine weit verbreitete Aktivierung der Gerinnungswege sowohl zu Thrombosen als auch zu Blutungen führt. Die Symptome sind sehr unterschiedlich, können aber Petechien, Ekchymosen, Blutungen aus Schleimhäuten, Organfunktionsstörungen und paradoxerweise auch thrombotische Ereignisse umfassen.
Erworbene Hämophilie: Die erworbene Hämophilie ist durch die Entwicklung von Autoantikörpern gegen Gerinnungsfaktoren, insbesondere Faktor VIII, gekennzeichnet. Die Patienten können spontane Blutungen in die Haut, die Muskeln oder die Weichteile sowie übermäßige Blutungen nach einem Trauma oder einer Operation aufweisen.
Komplikationen:
Die Komplikationen dieser Erkrankungen sind vielschichtig. Bei Hämophilie kann es zu Gelenkverformungen und chronischen Schmerzen kommen. Die Von-Willebrand-Krankheit kann zu wiederkehrenden, starken Blutungen führen, die möglicherweise eine Anämie verursachen. ITP und TTP können zu schweren inneren Blutungen führen, die lebenswichtige Organe angreifen. Die DIC kann zu Organversagen führen, während die erworbene Hämophilie lebensbedrohliche Blutungen verursachen kann, die ein wachsames Management und rasche Behandlungsmaßnahmen erfordern.
Untersuchung und Diagnose
Eine genaue Diagnose von Koagulopathien, Purpura und hämorrhagischen Störungen ist für eine wirksame Behandlung unerlässlich. Der diagnostische Prozess umfasst eine Kombination aus klinischer Untersuchung, Anamneseerhebung, speziellen Labortests und bildgebenden Untersuchungen.
Klinische Untersuchung:
Der diagnostische Weg beginnt in der Regel mit einer umfassenden Anamnese. Das medizinische Personal erkundigt sich nach den Symptomen des Patienten, einschließlich Blutungsneigung, leichten Blutergüssen und Anzeichen einer Anämie. Das Sammeln von Informationen über die Dauer und das Fortschreiten der Symptome ist für die Identifizierung möglicher Ursachen von entscheidender Bedeutung.
Nach der Erhebung der Krankengeschichte folgt eine gründliche körperliche Untersuchung. Der Arzt beurteilt die Vitalzeichen, das allgemeine Erscheinungsbild und spezifische Befunde im Zusammenhang mit Blutungsstörungen. Sie achten auf Purpura, Petechien, Ekchymosen, Gelenkdeformitäten, Organomegalie und Anzeichen einer Anämie. Besondere Aufmerksamkeit gilt den Schleimhäuten und dem Vorhandensein von Blutungen in Gelenke oder Weichteile.
Labortests und Bildgebung:
Vollständiges Blutbild (CBC): Ein vollständiges Blutbild ist ein grundlegender Bluttest, bei dem verschiedene Bestandteile des Blutes untersucht werden, darunter die Anzahl der roten Blutkörperchen, der Hämoglobinwert, der Hämatokritwert, die Anzahl der Blutplättchen und die Anzahl der weißen Blutkörperchen. Es wird verwendet, um Anämie, Thrombozytopenie oder Leukozytose festzustellen.
Gerinnungstests: Dieses Panel umfasst Tests wie die Prothrombinzeit (PT), die aktivierte partielle Thromboplastinzeit (aPTT) und das internationale normalisierte Verhältnis (INR). Diese Tests bewerten die Funktionalität der Gerinnungskaskade und können helfen, Anomalien bei den Gerinnungsfaktoren zu erkennen.
Thrombozytenzahl: Die Thrombozytenzahl bestimmt die Anzahl der Blutplättchen im Blut und hilft, eine Thrombozytopenie oder Thrombozythämie zu diagnostizieren, die für Blutungsstörungen relevant sind.
Peripherer Blutausstrich: Die mikroskopische Untersuchung eines peripheren Blutausstrichs ermöglicht die Beurteilung der Morphologie der roten Blutkörperchen, der Anomalien der Blutplättchen und des Vorhandenseins abnormaler Zellen, die auf bestimmte Erkrankungen hinweisen können.
Spezialisierte Gerinnungstests: Je nach klinischem Verdacht können spezifische Gerinnungsfaktortests durchgeführt werden, um Hämophilie oder die von-Willebrand-Krankheit zu diagnostizieren. Mit diesen Tests werden die Aktivitätswerte einzelner Gerinnungsfaktoren bestimmt.
Von-Willebrand-Faktor-Assay: Mit diesem Test werden das Antigen und die Aktivität des von-Willebrand-Faktors gemessen, was für die Diagnose der von-Willebrand-Krankheit unerlässlich ist.
Knochenmarkaspiration und -biopsie: In ausgewählten Fällen, in denen eine Beteiligung des Knochenmarks vermutet wird, kann eine Knochenmarkaspiration und -biopsie erforderlich sein. Dieses invasive Verfahren liefert entscheidende Erkenntnisse über das Ausmaß der Infiltration und ihre Auswirkungen auf die Hämatopoese.
Gentests: Gentests sind angezeigt, wenn der Verdacht auf erbliche Blutungsstörungen wie Hämophilie besteht. Sie können spezifische Genmutationen bestätigen, die für die Erkrankung verantwortlich sind.
Bildgebende Untersuchungen: Bildgebende Untersuchungen wie Ultraschall, Magnetresonanztomographie (MRT) oder Computertomographie (CT) können eingesetzt werden, um Gelenkblutungen, intrakranielle Blutungen oder andere innere Blutungsstellen zu untersuchen.
Die Kombination aus klinischer Untersuchung und gezielten Labor- und Bildgebungsuntersuchungen ermöglicht es den Ärzten, eine genaue Diagnose zu stellen, die maßgeschneiderte Behandlungsstrategien und eine umfassende Patientenversorgung ermöglicht.
Therapie und Behandlungen
Eine wirksame Behandlung von Koagulopathien, Purpura und hämorrhagischen Störungen konzentriert sich auf die Beseitigung der zugrunde liegenden Ursachen, die Behandlung der Symptome und die Verhinderung von Komplikationen. Der Behandlungsansatz kann je nach der spezifischen Erkrankung und ihrem Schweregrad variieren, wobei der Schwerpunkt auf individualisierten Behandlungsplänen liegt, die auf einer umfassenden klinischen Bewertung beruhen.
Hämostatische Therapien:
Transfusion von Blutbestandteilen: Bei schweren Blutungen können Transfusionen von Blutbestandteilen wie gepackten roten Blutkörperchen, Thrombozyten, gefrorenem Frischplasma und Kryopräzipitat verabreicht werden, um Mangelzustände zu korrigieren und die Gerinnungsfähigkeit zu verbessern.
Ersatz von Gerinnungsfaktoren: Bei Personen mit vererbten Gerinnungsstörungen wie Hämophilie oder von-Willebrand-Krankheit werden Gerinnungsfaktorkonzentrate verschrieben, um die Gerinnungsfunktion wiederherzustellen. Diese Konzentrate können prophylaktisch oder bei Bedarf infundiert werden.
Antifibrinolytische Wirkstoffe:
Tranexamsäure und Aminocapronsäure: Diese Medikamente hemmen die Fibrinolyse, d. h. den Abbau von Blutgerinnseln. Sie werden häufig zur Behandlung von Blutungsepisoden bei Personen mit Blutungsstörungen eingesetzt.
Thrombozytenfördernde Mittel:
Thrombopoietin-Rezeptor-Agonisten: Bei Thrombozytopenie stimulieren diese Medikamente die Thrombozytenproduktion im Knochenmark und können zur Erhöhung der Thrombozytenzahl eingesetzt werden.
Von-Willebrand-Faktor-Ersatz:
Desmopressin: Dieses Medikament kann die Freisetzung des gespeicherten von-Willebrand-Faktors anregen, insbesondere in leichten Fällen der von-Willebrand-Krankheit.
Immunmodulatoren:
Immunsuppressiva: Bei Autoimmunerkrankungen, die mit Thrombozytopenie oder anderen Blutungsproblemen einhergehen, können immunsuppressive Medikamente eingesetzt werden, um die Aktivität des Immunsystems zu verringern und die Krankheit zu kontrollieren.
Hämatopoetische Stammzelltransplantation:
Knochenmark- oder Stammzelltransplantation: Diese Behandlungsoption ist schweren angeborenen Gerinnungsstörungen oder Knochenmarksstörungen vorbehalten. Dabei wird das kranke Knochenmark durch gesunde Stammzellen ersetzt, um die normale Blutzellproduktion wiederherzustellen.
Chirurgische Eingriffe:
Gelenkdrainage und Operation: Bei Personen mit wiederkehrenden Gelenkblutungen kann eine Gelenkaspiration oder eine Operation erforderlich sein, um angesammeltes Blut zu entfernen und Gelenkschäden zu verhindern.
Symptomatische Behandlung:
Schmerzbehandlung: Analgetika und physikalische Therapie können eingesetzt werden, um die Schmerzen zu lindern und die Gelenkfunktion bei Personen mit Gelenkblutungen zu verbessern.
Unterstützende Pflege:
Psychosoziale Unterstützung: Der Umgang mit chronischen Blutungsstörungen kann psychosoziale Unterstützung und Beratung erfordern, um die emotionalen und psychischen Aspekte der Erkrankung zu behandeln.
Behandlung der zugrunde liegenden Erkrankungen:
Behandlung von Grunderkrankungen: Wenn Gerinnungsprobleme sekundär zu einer Grunderkrankung auftreten, ist die Behandlung der Grunderkrankung ein wichtiger Teil der Behandlung.
Es ist wichtig zu betonen, dass die Behandlung dieser Störungen sehr individuell ist. Die Gesundheitsdienstleister arbeiten eng mit den Patienten zusammen, um die Behandlungspläne auf ihre spezifischen Bedürfnisse abzustimmen, wobei Faktoren wie Art und Schweregrad der Störung, das Vorhandensein von Komplikationen und der allgemeine Gesundheitszustand des Patienten berücksichtigt werden.
Ursachen und Risikofaktoren
Das Verständnis der Ursachen und Risikofaktoren, die mit Koagulopathien, Purpura und hämorrhagischen Störungen einhergehen, ist sowohl für die Prävention als auch für eine wirksame Behandlung unerlässlich. Dieser Abschnitt befasst sich mit den zugrunde liegenden biologischen Prozessen, die diese Erkrankungen verursachen, und zeigt die damit verbundenen Risikofaktoren auf.
Ursachen:
Koagulopathien, Purpura und hämorrhagische Störungen entstehen durch Störungen in den komplexen Prozessen der Blutgerinnung und der Thrombozytenfunktion. Auf biologischer Ebene können diese Erkrankungen auf genetische Mutationen, Autoimmunreaktionen oder erworbene Störungen zurückgeführt werden.
Vererbte Genmutationen spielen bei der Entstehung dieser Erkrankungen eine wichtige Rolle. Hämophilie beispielsweise entsteht durch Mutationen in Genen, die für die Produktion der Gerinnungsfaktoren VIII oder IX verantwortlich sind. Diese Mutationen führen zu einer verminderten oder fehlenden Aktivität der Gerinnungsfaktoren, wodurch die Fähigkeit des Blutes, stabile Gerinnsel zu bilden, beeinträchtigt wird. Auch die von-Willebrand-Krankheit, die häufigste vererbte Blutungsstörung, beruht auf Mutationen, die das Gen für den von-Willebrand-Faktor betreffen, wodurch die Adhäsion der Blutplättchen und die Gerinnungsbildung gestört werden.
Auch Autoimmunreaktionen können zu Gerinnungsstörungen führen. Die immunvermittelte thrombozytopenische Purpura (ITP) tritt auf, wenn sich das Immunsystem gegen Blutplättchen richtet und diese zerstört, wodurch die Gerinnungsbildung beeinträchtigt wird und es zu Blutungsepisoden kommt. In einigen Fällen können sich Autoantikörper gegen Gerinnungsfaktoren richten, wie dies bei der erworbenen Hämophilie der Fall ist.
Erworbene Koagulopathien können die Folge von Grunderkrankungen sein. So können beispielsweise Lebererkrankungen die Produktion von Gerinnungsfaktoren stören, was zu einer Blutungsneigung führt. Außerdem können bestimmte Medikamente wie Antikoagulanzien oder Thrombozytenaggregationshemmer die Blutgerinnung beeinträchtigen und so das Blutungsrisiko erhöhen.
Risikofaktoren:
Familienanamnese: Personen mit einer familiären Vorgeschichte von Koagulopathien oder Blutungsstörungen können eine genetische Prädisposition für diese Erkrankungen haben, was ihr Risiko erhöht.
Geschlecht: Einige Blutungsstörungen, wie z. B. die Hämophilie, betreffen aufgrund ihres X-chromosomalen Vererbungsmusters häufiger Männer.
Alter: Altersbedingte Faktoren, wie die Entwicklung von immunvermittelten Erkrankungen wie ITP bei Kindern, können die Anfälligkeit beeinflussen.
Medikamente: Die Einnahme von Antikoagulanzien, Thrombozytenaggregationshemmern oder Medikamenten, die die Blutgerinnung beeinflussen, kann das Risiko von Blutungsepisoden erhöhen.
Grundlegende Gesundheitszustände: Chronische Lebererkrankungen, Autoimmunerkrankungen und bestimmte bösartige Erkrankungen können das Risiko für erworbene Koagulopathien erhöhen.
Trauma oder Operation: Physische Traumata oder chirurgische Eingriffe können bei Personen mit zugrunde liegenden Gerinnungsstörungen Blutungsepisoden auslösen.
Schwangerschaft: Hämorrhagische Störungen wie die Schwangerschaftsthrombozytopenie können während der Schwangerschaft auftreten und das Risiko von Blutungskomplikationen erhöhen.
Infektionen: Einige Infektionen, wie z. B. Hepatitis, können die Leberfunktion beeinträchtigen und zu Gerinnungsstörungen beitragen.
Lebensstil-Faktoren: Faktoren wie übermäßiger Alkoholkonsum oder eine Ernährung mit einem Mangel an wichtigen Nährstoffen können die Blutgerinnung beeinträchtigen.
Es ist wichtig zu beachten, dass diese Risikofaktoren zwar die Wahrscheinlichkeit der Entwicklung von Koagulopathien, Purpura und hämorrhagischen Störungen erhöhen können, ihr Vorhandensein aber keine Garantie für die Entwicklung dieser Erkrankungen ist und umgekehrt.
Krankheitsverlauf und Prognose
Das Verständnis des Krankheitsverlaufs von Koagulopathien, Purpura und hämorrhagischen Störungen sowie deren Prognose ist für Patienten und Gesundheitsdienstleister von entscheidender Bedeutung. In diesem Abschnitt erfahren Sie, wie sich diese Erkrankungen typischerweise entwickeln und welche Folgen sie haben können.
Krankheitsverlauf:
Der Verlauf von Koagulopathien, Purpura und hämorrhagischen Störungen kann je nach der zugrunde liegenden Ursache und individuellen Faktoren variieren. Es gibt jedoch gemeinsame Muster für den typischen Verlauf dieser Erkrankungen.
Im Allgemeinen können sich die Symptome allmählich oder plötzlich manifestieren, je nach Subtyp und Schweregrad. Die Diagnose wird in der Regel gestellt, wenn bei den Betroffenen Blutungsepisoden auftreten, z. B. leichte Blutergüsse, anhaltende Blutungen bei kleineren Verletzungen oder spontanes Nasen- oder Zahnfleischbluten. Diagnostische Tests, darunter Blutuntersuchungen und spezielle Tests, helfen dabei, die spezifische Störung zu bestätigen.
Nach der Diagnose variiert der Verlauf je nach der zugrunde liegenden Ursache. Bei vererbten Gerinnungsstörungen wie der Hämophilie ist der Verlauf im Laufe der Zeit oft stabil, und die Symptome werden durch geeignete Behandlungen kontrolliert. Im Gegensatz dazu können erworbene Koagulopathien dem Verlauf der Grunderkrankung folgen. Wenn die Grunderkrankung behandelt wird, kann sich die Blutungsneigung verbessern.
Die Komplikationen im Zusammenhang mit diesen Störungen können vielfältig sein. Anhaltende Blutungsepisoden können zu Anämie und in schweren Fällen zu Organschäden führen. Außerdem besteht bei Menschen mit Gerinnungsstörungen die Gefahr, dass sie bei Operationen oder traumatischen Ereignissen übermäßig bluten. Daher ist eine rasche Behandlung von Blutungsepisoden entscheidend, um weitere Komplikationen zu verhindern.
Prognose:
Die Prognose für Personen mit Koagulopathien, Purpura und hämorrhagischen Störungen hängt von verschiedenen Faktoren ab.
Der spezifische Subtyp und dessen Schweregrad beeinflussen die Prognose erheblich. Einige Erkrankungen, wie z. B. die leichte von-Willebrand-Krankheit, können bei entsprechender Behandlung zu einer nahezu normalen Lebenserwartung führen. Im Gegensatz dazu kann eine schwere Hämophilie zu häufigeren und schwereren Blutungsepisoden führen, die das allgemeine Wohlbefinden beeinträchtigen.
Eine rechtzeitige und wirksame Behandlung hat einen erheblichen Einfluss auf die Prognose. Patienten, die gut auf Therapien wie den Ersatz von Gerinnungsfaktoren oder thrombozytenfördernden Medikamenten ansprechen, können ein relativ normales Leben mit minimalen Komplikationen führen.
Das Vorhandensein und die Behandlung von Komplikationen sind entscheidende Faktoren für die Prognose. Eine frühzeitige Behandlung von blutungsbedingten Komplikationen kann langfristige Organschäden verhindern und die Gesamtergebnisse verbessern.
Individuelle Patientenfaktoren wie die Einhaltung von Behandlungsschemata, die Wahl des Lebensstils und der Zugang zur medizinischen Versorgung spielen eine wichtige Rolle für die Prognose. Patienten, die sich aktiv an ihrer Behandlung beteiligen, haben tendenziell bessere Ergebnisse.
Prävention
Die Vorbeugung von Koagulopathien, Purpura und hämorrhagischen Störungen ist wichtig, um das Risiko dieser Erkrankungen und der damit verbundenen Komplikationen zu verringern. Dieser Abschnitt befasst sich mit verschiedenen Präventionsmethoden, einschließlich Strategien zur Minimierung des Risikos, an diesen Erkrankungen zu erkranken, und Maßnahmen zur Minderung des Risikos von Blutungsepisoden.
Genetische Beratung:
Für Personen mit einer familiären Vorbelastung durch erbliche Gerinnungsstörungen ist die genetische Beratung eine wertvolle Hilfe. Sie hilft den Betroffenen, ihr Risiko zu verstehen und fundierte Entscheidungen über die Familienplanung zu treffen.
Frühzeitige Diagnose und Behandlung:
Eine rechtzeitige Diagnose und eine angemessene Behandlung sind entscheidend für die Vermeidung von Komplikationen im Zusammenhang mit vererbten Gerinnungsstörungen. Regelmäßige Untersuchungen und Kontrollen können helfen, Blutungsneigungen frühzeitig zu erkennen und zu behandeln.
Behandlung der zugrunde liegenden Erkrankung:
In Fällen, in denen Koagulopathien sekundär zu anderen Erkrankungen auftreten, ist eine wirksame Behandlung der Grunderkrankung von entscheidender Bedeutung. Dies kann die Kontrolle chronischer Krankheiten, Infektionen oder entzündlicher Störungen beinhalten, um das Risiko von Blutungsepisoden zu verringern.
Überprüfung der Medikation:
In einigen Fällen können Gerinnungsstörungen durch Medikamente ausgelöst oder verschlimmert werden. Gesundheitsdienstleister können die Notwendigkeit von Medikamenten prüfen, alternative Behandlungsmöglichkeiten erforschen oder die Dosierung anpassen, wenn dies möglich ist, um das Blutungsrisiko zu verringern.
Arzneimittelsicherheit:
Um arzneimittelinduzierten Koagulopathien vorzubeugen, sollten Gesundheitsdienstleister der Arzneimittelsicherheit Vorrang einräumen. Sie können potenzielle Risiken und Vorteile abwägen und alternative Medikamente oder Behandlungsmöglichkeiten in Betracht ziehen, die die normalen Gerinnungsmechanismen weniger stark beeinträchtigen.
Patientenaufklärung:
Die Aufklärung der Patienten über mögliche Nebenwirkungen von Medikamenten und die Wichtigkeit, ungewöhnliche Symptome sofort zu melden, kann dazu beitragen, schwere Fälle von arzneimittelinduzierten Koagulopathien zu verhindern.
Pränataldiagnose:
Für werdende Eltern, in deren Familie diese Störungen vorkommen, kann die Pränataldiagnose wertvolle Informationen über den Gesundheitszustand des Fötus liefern. Anhand dieser Informationen können Entscheidungen über das Schwangerschaftsmanagement und bei Bedarf über frühzeitige Interventionen getroffen werden.
Änderungen der Lebensweise:
Bei Personen mit Gerinnungsstörungen können bestimmte Änderungen des Lebensstils dazu beitragen, das Risiko von Blutungsepisoden zu minimieren. Dazu kann es gehören, risikoreiche Aktivitäten oder Sportarten zu vermeiden, die zu Verletzungen führen könnten, und Vorsichtsmaßnahmen zu treffen, um Unfälle zu vermeiden.
Bereitschaft für den Notfall:
Personen mit bekannten Gerinnungsstörungen sollten auf Notfälle gut vorbereitet sein. Sie sollten medizinische Warnhinweise bei sich tragen, vor medizinischen Eingriffen das medizinische Personal über ihren Zustand informieren und Zugang zu notwendigen Gerinnungsfaktorbehandlungen oder Medikamenten haben.
Regelmäßige Nachsorge:
Regelmäßige Nachsorgetermine bei Gesundheitsdienstleistern sind für Menschen mit Koagulopathien unerlässlich. Dadurch wird sichergestellt, dass die Behandlungspläne auf dem neuesten Stand sind und Blutungen wirksam verhindert werden.
Es ist wichtig zu wissen, dass nicht alle Fälle von Koagulopathien, Purpura und hämorrhagischen Störungen vermeidbar sind, insbesondere wenn sie auf vererbte genetische Faktoren zurückzuführen sind. Eine frühzeitige Diagnose und eine wirksame Behandlung können jedoch das Risiko von Komplikationen erheblich verringern und die Lebensqualität der Betroffenen verbessern.
Zusammenfassung
Koagulopathien, Purpura und hämorrhagische Störungen bilden eine Gruppe von Blutkrankheiten, die durch eine abnorme Blutungsneigung gekennzeichnet sind. Zu den Symptomen gehören ungeklärte Blutungen, Blutergüsse, Petechien und anhaltende Blutungen bei kleineren Verletzungen. Die Prävalenz dieser Erkrankungen ist weltweit unterschiedlich und das Verständnis hat sich im Laufe der Zeit weiterentwickelt. Die Komplikationen können schwerwiegend sein und zu Blutarmut und Organschäden führen. Die genaue Diagnose erfordert eine klinische Beurteilung, eine Anamnese und spezielle Tests. Die Behandlungsstrategien sind auf die jeweilige Person zugeschnitten und gehen sowohl auf die zugrunde liegenden Ursachen als auch auf die Symptome ein. Diese Störungen resultieren aus einer Störung der Blutgerinnung und der Thrombozytenfunktion, die häufig durch genetische Mutationen, Autoimmunreaktionen oder erworbene Faktoren beeinflusst wird. Zu den Risikofaktoren gehören die Familienanamnese, das Geschlecht, das Alter, Medikamente, Grunderkrankungen, Traumata und der Lebensstil. Um das Risiko und die Komplikationen zu minimieren, sollten Sie eine genetische Beratung, eine frühzeitige Diagnose, eine wirksame Behandlung der Grunderkrankungen, die Sicherheit von Medikamenten, Aufklärung, pränatale Vorsorgeuntersuchungen, Anpassungen des Lebensstils, Vorsorge für Notfälle und eine regelmäßige Nachsorge durch die Gesundheitsdienstleister anstreben.