

Die Polyarteriitis Nodosa (PAN) ist eine Vaskulitis mittlerer und kleinerer Gefässe. Die Inzidenz wird auf 1,6 bis 1,9 Fälle pro Millionen Einwohner pro Jahr geschätzt, was ein Grund dafür sein dürfte, dass die Leitlinien seit geraumer Zeit nicht mehr aktualisiert wurden und es teilweise neue Erkenntnisse gibt, die noch keinen Eingang gefunden haben. Eine Expertin gab auf dem Kongress der DGRh Überblick über die aktuelle Diagnostik und Therapie.
Ein Grund für den statistisch signifikanten Rückgang der PAN-Fälle in den vergangenen Jahren dürften die verbesserten Behandlungsmöglichkeiten bei Hepatitis B sein, erklärte Prof. Dr. Ina Kötter, Klinik für Rheumatologie und Immunologie, Sektion für Rheumatologie und entzündliche Systemerkrankungen des UKE, Klinikum Bad Bramstedt [1]. Bei einer Prävalenz von 30 pro Millionen Einwohner sei es aber auch nicht verwunderlich, dass es nicht viele Studien zu der Thematik gebe. «Es ist ein Problem, dass anders als bei anderen Vaskulitiden keine neuen Klassifikations- und Diagnosekriterien existieren, sondern immer noch die alten ACR-Vorgaben von 1990 gelten.» Denn einerseits stehe dort die Hepatitis B als Auslöser, was heute kaum mehr der Fall sei. Zweitens werde die Nierenbeteiligung definiert als erhöhter Serumharnstoff oder Kreatinin. «Wenn das so weit ist, kommt man natürlich schon zu spät», so die Rheumatologin.
Die Kinderrheumatologen waren diesbezüglich schneller und haben 2010 in den EULAR/PRINTO-Kriterien für die pädiatrische PAN die Nierenbeteiligung deutlich sensitiver definiert und die Proteinurie mit einbezogen. Auch kommt die Hepatitis hier nicht mehr vor. Prof. Kötters Einschätzung nach werde es in Zukunft auch in die Richtung eines Punktesystems gehen, bei dem Faktoren wie Mononeuritis multiplex, gastrointestinale Symptomatik, Abwesenheit von MPO-ANCA, Proteinurie, Allgemeinsymptomatik, angiografische Abnormitäten, also multiple aneinander geschaltete Aneurysmen, und auch die typische Histologie jeweils mit einem Punkt zählen und die Kriterien bei Erreichen einer bestimmten Gesamtpunktzahl erfüllt sind.
Bzgl. der Symptomatik weisen viele Patienten Arthralgien und über 60% auch Myalgien auf. Auch die Neuropathien, ZNS-Beteiligung, Hautveränderungen und Nierenbeteiligung sind relativ häufig. Einen wesentlichen Unterschied gibt es zwischen Kindern und Erwachsenen: Die Erwachsenen haben deutlich mehr periphere neurologische Beteiligung (PNS), Kinder dafür mehr ZNS-Beteiligung. «Dazu muss man noch im Hinterkopf haben, dass eine kindliche PAN immer verdächtig ist auf eine Adenosin-Deaminase-2-Defizienz (Deficiency of adenosine deaminase 2, DADA2), eine genetisch bedingte Vaskulitis mit im Vordergrund stehender ZNS-Beteiligung, aber auch mit einem Immundefekt.»
Koronariitis – das vergessene Symptom
In einer retrospektiven chinesischen Analyse mit 145 PAN-Patienten hatten 13,7% (n=19) Koronararterien-Aneurysmen und daraus resultierend eine KHK gefunden hat. Bei der Auswertung hat man gesehen, dass die Patienten mit Aneurysmen weniger Gewichtsverlust hatten, weniger subkutane und mehr periphere Gefässbeteiligung, vor allem kranielle Arterien, Carotis, Niere, Darm und untere Extremitäten. Auch Hypertonie trat verstärkt auf.
Wenn jemand bei einer KHK keine typischen kardiovaskuläre Faktoren hat, dann sollte man auf jeden Fall nach weiterer Organbeteiligung schauen, Antikörper untersuchen, Entzündungsparameter checken und im Zweifel auch den Rheumatologen hinzuziehen, lautete die Schlussfolgerung. In der Multivarianzanalyse zeigte sich, dass v.a. die Beteiligung der Darmarterien (OR 3,722; p=0,033) und die Hypertonie (OR 6,668; p=0,003) Risikofaktoren für eine Koronararterien-Beteiligung waren. «Wenn man Patienten mit einer PAN und diesen Symptomen hat, dann sollte man dezidiert nach einer Koronar-Beteiligung suchen, bevor die Patienten diesbezüglich symptomatisch werden», so der Rat von Prof. Kötter.
Ältere sind häufiger systemisch
Ein Blick auf die Altersverteilung zeigt, dass Patienten mit einem milden kutanen Krankheitsbild jünger sind, solche mit schwerer kutaner PAN etwas älter, und Menschen mit der systemischen PAN haben das höchste Lebensalter.
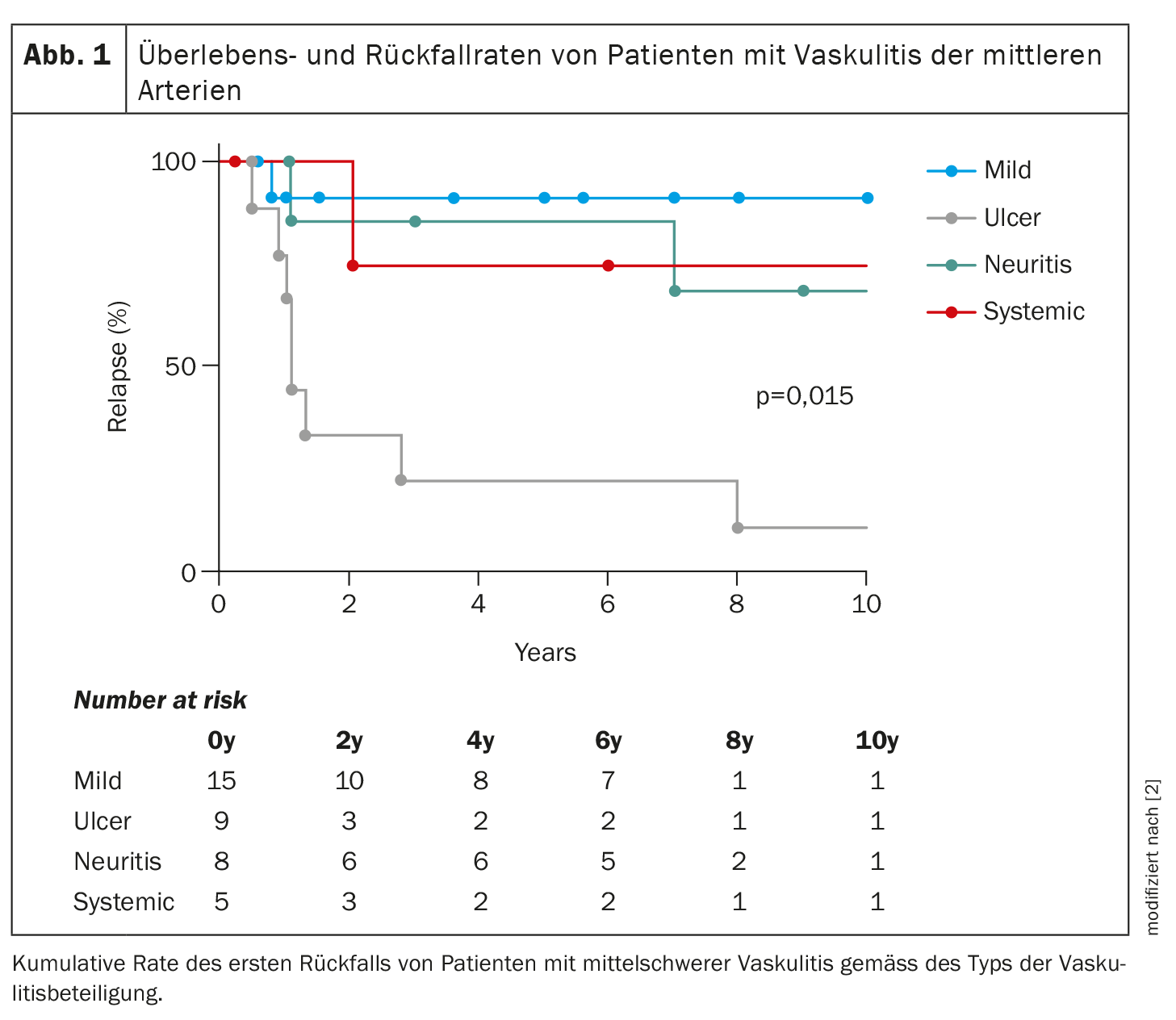
Bezogen auf das Langzeitüberleben weist die systemische PAN die schlechtesten Werte auf, «was aber spannend ist: Die häufigsten Rezidive mit Ulzera treten bei der schweren kutanen PAN treten auf», sagte die Expertin (Abb. 1). Bei der systemischen PAN kommt es also zu weniger Rezidiven als bei der schweren kutanen PAN.
Pathogenetisch geht man von Umwelttriggern aus. Zusätzlich gibt es Komorbiditäten wie die chronische myelomonozytäre Leukämie (CMML). Auch wird eine immungenetische Prädisposition vermutet. Tatsächlich, so Prof. Kötter, sei dies aber bei der PAN im Gegensatz zu anderen Vaskulitiden noch gar nicht richtig definiert und untersucht.
Diagnostik und Therapie
Nicht jeder PAN-Patient muss an der Haut betroffen sein. Neben der Hautbiopsie stehen diagnostisch noch weitere Methoden zur Verfügung: Bei muskulärer PAN stehen MRT und Ultraschall zur Verfügung. Dort werden Hypo- und Hyperechogenitäten sichtbar, die dann mit der Entzündung im MRT korrelieren. Dazu gibt es das PET-CT: Eine aktuelle Arbeit aus Frankreich hat 10 Patienten untersucht, die alle Myalgien, Arthralgien und systemische Symptome hatten und bei denen die PAN bereits hautbioptisch gesichert war. Bei allen wurden Anreicherungen im PET-CT gefunden, entweder im Muskel, linear oder fokal, und bei 4 von 10 auch in den grossen Gefässen.
Im Gegensatz zu den Diagnosekriterien gibt es zur Therapie der PAN Empfehlungen des American College of Rheumatology (ACR). Bei aktiver Erkrankung wird dort zu i.v. Cyclophosphamid geraten plus Methylprednisolon. Das Cyclophosphamid soll bis zur Remission beibehalten werden, dann folgt eine Umstellung auf ein anderes Immunsuppressivum, vorzugsweise MTX, AZA oder MMF. Wenn keine schwere Organbeteiligung vorliegt, werden die Immunsuppressiva direkt plus Glukokortikoid eingesetzt. Bei Refraktärität wird eine Eskalation auf die vorgenannte Therapie angeraten.
«Was nicht in den ACR-Empfehlungen vorkommt, ist die DADA2-Defizienz – diese wird direkt mit einem TNF-Inhibitor bekämpft.» Ob man die auf die Haut limitierte PAN nur mit Hydroxychloroquin behandeln kann, zog Prof. Kötter in Zweifel, «und was auch eine wesentliche Überlegung sein muss: Was machen wir denn, wenn Cyclophosphamid nicht hilft?» Für diesen Fall verwies sie auf eine retrospektive Arbeit aus Frankreich, in der Biologika miteinander verglichen wurden, wenn auch nur in einer kleinen Kohorte zwischen 10 und 18 Patienten.
Bei der Gegenüberstellung Rituximab vs. TNF-Blocker vs. Tocilizumab verhalf Tocilizumab zu den Remissionen, verursachte aber auch die meisten Nebenwirkungen, darunter zum Teil auch intestinale Perforationen. Der BVAS als Aktivitätsscore ging mit Tocilizumab nach 12 Monaten sogar auf 0, und auch in der Kaplan-Meier Kurve schien der Wirkstoff den anderen Substanzen überlegen zu sein.
Eine indische Untersuchung zu JAK-Inhibitoren schloss vier Fälle mit kutaner PAN ein und wies Verbesserungen auf. Das könne vielleicht ein weiteres vielversprechendes Therapieprinzip sein, schloss die Referentin.
Quelle:
- Kötter I: Vortrag «Seltene Vaskulitiden»; Deutscher Rheumatologiekongress 2023, Leipzig, 31.08.2023.
- Shirai T, Shirota Y, Fujii H, et al.: Four distinct clinical phenotypes of vasculitis affecting medium-sized arteries. Scandinavian Journal of Rheumatology 2019; 48: 308-314; doi: 10.1080/03009742.2018.1551965.
InFo RHEUMATOLOGIE 2023; 5(2):26–27
Autoren
- Jens Dehn
Publikation
- INFO RHEUMATOLOGIE

Comments are closed.