

Die innovativen systemischen Behandlungsmöglichkeiten der Psoriasis sind ein Paradebeispiel für die translationale Medizin. In einem im vergangenen Jahr im Journal of Investigative Dermatologyerschienenen Review gibt Prof. Dr. med. Peter van de Kerkhof, ehemaliger Präsident der European Society for Dermatological Research (ESDR) und Chief Medical Officer beim International Psoriasis Council, einen aktuellen Überblick zu therapierelevanten immunpathogenetischen Konzepten der Psoriasis.
In den letzten Dekaden wurden beachtliche Fortschritte in der Aufklärung der komplexen und multifaktoriellen Pathogenese der Psoriasis erzielt als Basis für die Entwicklung hochwirksamer Therapieoptionen [1]. Heute gilt auf zellulärer und molekularer Ebene eine T-Zell-vermittelte Immunreaktion und ein damit einhergehendes charakteristisches Zytokinmilieu als wesentlicher Pathomechanismus [2]. Auf genetischer Ebene wurden zahlreiche mit Psoriasis assoziierten Genloci identifiziert. Als Hauptrisikoallel der Psoriasis vulgaris gilt HLA-C*06:02 [3]. Dieses befindet sich auf dem Psoriasis-Suszeptibilitätslokus PSORS1. Einen Beleg für autoimmune Pathomechanismen bei Psoriasis lieferte der Befund, dass HLA-C*06:02 eine Autoimmunantwort gegen ADAMTSL5, ein melanozytäres Autoantigen, hervorrief [17,18]. Neben ADAMTSL5 sind LL-37/Cathelicidin und PLA2G4D zwei weitere potenzielle Autoantigene, welche für die Initiierung oder Aufrechterhaltung psoriatischer Läsionen eine Rolle zu spielen scheinen [19].
Psoriasis als T-Zell-vermittelte Autoimmunerkrankung
An der Immunpathogenese der Psoriasis sind Prozesse des angeborenen und des erworbenen Immunsystems beteiligt (Abb. 1). In den Beginn der Entzündungskaskade involviert sind Zellen des angeborenen Immunsystems. Dabei scheinen dendritische Zellen (DC) und Makrophagen eine Schlüsselrolle zu spielen in der Etablierung einer von den T-Helferzellen-1 (Th1) und Th17 dominierten Immunantwort. Die Aktivierung von DC in der Haut führt zu einer Freisetzung der Interleukine-12 (IL-12) und IL-23, welche die naiven T-Zellen zur Differenzierung in Th1- (durch IL-12) und Th17-Zellen (durch IL-23) anregen [4]. T-Helfer (Th) -Zellen koordinieren die zellvermittelte adaptive Immunantwort. Aktivierte Th1- und Th17-Zellen sezernieren die Zytokine IFN-γ und TNF-α bzw. IL-17 und IL-22, welche die weitere Pathogenese vorantreiben [5,6].
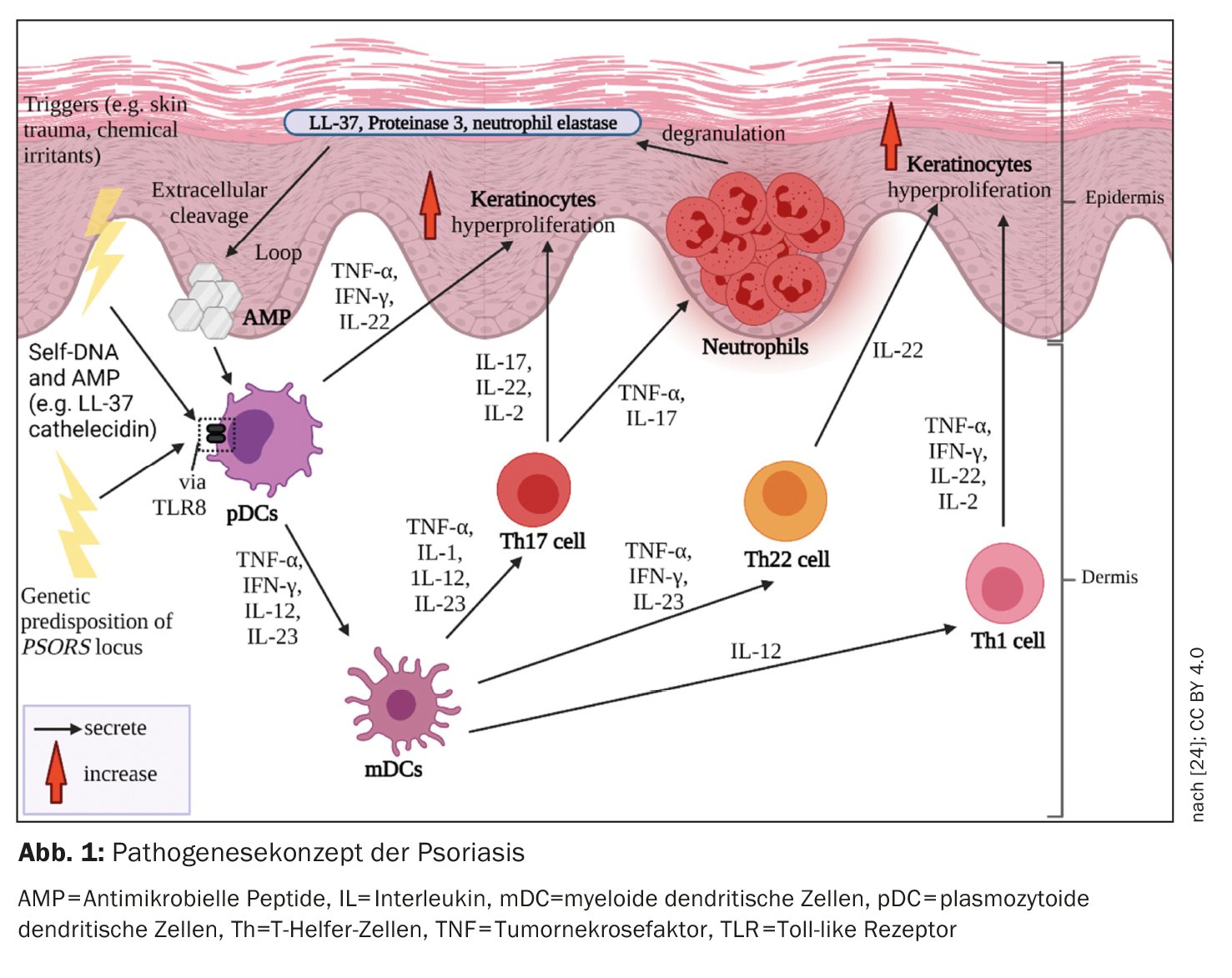
Proinflammatorisches Milieu
Bei Psoriasis wird die Immunantwort hauptsächlich durch Th1-Zellen vermittelt. wohingegen bei atopischer Dermatitis eine Th2-Dominanz vorliegt. T-Zellpopulationen in Biopsien von psoriatischen Läsionen, welche im Vergleich zu Läsionen im Rahmen von atopischer Dermatitis beim selben Patienten unterschiedliche T-Zellinfiltrate zeigten, bestätigen das Th1/Th2-Paradigma [1,8]. TNF-α, IFN-γ, IL-17A, IL-17F, IL-22, und IL-6 bilden in psoriatischen Läsionen ein Zytokinnetzwerk, das unter anderem Hyperproliferation der Keratinozyten induziert [1]. Der beschleunigte Differenzierungsvorgang der Keratinozyten verkürzt deren Transitzeit vom Stratum basale bis zum Stratum corneum von etwa 30 auf 5–8 Tage [9]. Dies resultiert in der für Psoriasis typischen Schuppung und Verdickung der Epidermis. Aktivierte Keratinozyten produzieren ebenfalls proinflammatorische Mediatoren und exprimieren Adhäsionsmoleküle auf der Zelloberfläche, was zu einer Verstärkung und Chronifizierung der Immunantwort führt [10].
Immunologisches Gedächtnis: «Tissue Resident Memory T cells»
Psoriatische Läsionen weisen eine höhere Gesamtzellzahl an Immunzellen auf im Vergleich zu gesunder Haut [11]. Auch in der Haut von Psoriasis-Patienten, die klinisch keine Läsionen zeigt, liess sich eine höhere Anzahl T-Zellen als in gesunder Haut nachweisen [11]. Intraepidermale Akkumulation von CD8-Zellen ist entscheidend für die Entwicklung psoriatischer Läsionen [1]. Aber wie kommt es dazu? Nachdem Autoantigene den dendritischen Zellen präsentiert wurden, kommt es zu einer T-Zellaktivierung. Aktivierte T-Zellen wandern in die Epidermis ein, wobei komplexe Vorgänge notwendig sind, um in die Epidermis zu gelangen – man vermutet, dass α1β1-Integrine dabei eine wichtige Rolle spielen. Die Blockade von α1β1 durch einen monoklonalen Antikörper resultierte im Mausmodell in der Akkumulation epidermaler T-Zellen und in der Entwicklung psoriatischer Läsionen [12].
T-Zellen, die sich in der Epidermis befinden, spielen bei der Pathogenese der Psoriasis eine äusserst wichtige Rolle. In psoriatischen Plaques finden sich CD4-positive Zellen vor allem in der Dermis und CD8-positive Zellen überwiegend in der Epidermis [13]. Ein grosser Anteil an CD8-positiven Zellen in der Epidermis zählen zu den «Tissue Resident Memory T cells» (TRM Zellen, CD8+ TRM). Hierbei handelt es sich um Gedächtnis-T-Zellen, die in abgeklungenen psoriatischen Läsionen verbleiben und ein immunologisches Gedächtnis aufrechterhalten, was erklärt, weshalb Psoriasisläsionen oft an denselben Stellen wiederauftreten [14]. CD8+ TRM sind ein potenzieller Biomarker für eine residuale Krankheitsaktivität [15,16].
Medizinhistorischer Rückblick: antipsoriatische Systemtherapien
In den 1980-er Jahren bestätigten Untersuchungen mit dem Immunsuppressivum Ciclosporin A, das sich in der Behandlung der Psoriasis als wirksam erwies, dass es sich um eine T-Zell-vermittelte Erkrankung handelt [26,27]. Ciclosporin hemmt im Zytoplasma die Phosphatase Calcineurin und blockiert dadurch die Phosphorylierung des nukleären Faktors für T-Zellen (NFATc), welcher zur Aktivierung der Gene für verschiedene proinflammatorische Zytokine zuständig ist [1]. Eine weitere Untermauerung der These, dass T-Zellen ein wichtiges therapeutisches Target sind, lieferten die antipsoriatischen Effekte von anti-CD4 und CTLA-4–Ig [28,29]. Eine US-amerikanische Arbeitsgruppe zeigte in den 1990-er Jahren, dass durch das Einspritzen autologer T-Zellen bei auf SCID-Mäusen transplantierter, nicht-erkrankter psoriatischer Haut Läsionen induziert werden konnten [30]. Im gleichen Modell konnte gezeigt werden, dass CD4+ T-Lymphozyten psoriatische Läsionen in nicht-erkrankter psoriatischer Haut induzieren [31].
| Monoklonale Antikörper und «Small Molecules» Biologika haben die Behandlungsmöglichkeiten der Psoriasis revolutioniert. Mit den heute verfügbaren biologischen Wirksubstanzen kann eine langfristig wirksame und sichere Krankheitskontrolle erreicht werden. Während durch den Einsatz von anti-TNF-α-Antikörpern bei einer Mehrheit der Patienten PASI75#erzielt werden kann, führen anti-IL17A-Antikörper und anti-IL-23 Antikörper bei rund der Hälfte der Patienten zu einem PASI100**, was die Relevanz des IL-23/IL-17-Signalwegs bei Psoriasis verdeutlicht. Die Wirksamkeit von Biologika wurde in Netzwerk-Metaanalysen verglichen [20]. Je nach Patientenmerkmalen, einschliesslich eventuell vorliegender Komorbiditäten, ist eher die eine oder die andere Substanzklasse vorzuziehen. In der Schweiz sind derzeit neben den TNF-α-Hemmern und dem IL12-/23-Antikörper Ustekinumab, mehrere Wirksubstanzen aus den Gruppen der anti-IL17A-Antikörper (Secukinumab, Ixekizumab) und der anti-IL-23 Antikörper (Guselkumab, Risankizumab, Tildrakizumab) zugelassen [25]. Und auch der IL17A/17F-Inhibitor Bimekizumab hat inzwischen die Zulassungshürden überwunden [25]. Aus dem Bereich der «Small Molecules» ist der PDE-4-Hemmer Apremilast ein zugelassenes orales Medikament. Daneben werden aktuell Deucravacitinib (oraler TYK2-i), Roflumilast (topischer PDE-4-i) und Tapinarof (topischer AHR-Agonist) klinisch geprüft [21–23]. |
| # PASI75 = eine Verbesserung des PASI (Psoriasis Area and Severity Index) um mindestens 75% ** PASI 100 = eine Verbesserung des PASI (Psoriasis Area and Severity Index) um 100% |
CD4+ Th-Zellen sezernieren bekanntlich verschiedene Zytokine. Die Bedeutung dieser Botenstoffe in der Psoriasis-Pathogenese wird bestätigt durch das sehr gute therapeutische Ansprechen bei gezielter Neutralisation einzelner Zytokine des Immunsystems. Zunächst stellte man fest, dass die Blockade von TNF-α zur Linderung der Psoriasis beiträgt [32,33]. Mit Ustekinumab folgte später die Zulassung eines Biologikums, das sich gegen die gemeinsame p40-Untereinheit von IL-12 und IL-23 richtet und eine gute Wirksamkeit bei Psoriasis zeigte [1]. Als noch effektiver erwiesen haben sich monoklonale Antikörper, welche selektiv auf die p19-Untereinheit von IL-23 abzielen (Guselkumab, Risankizumab, Tildrakizumab) (Kasten). Und ebenfalls hochwirksam sind Biologika, die sich gegen IL-17A richten (Secukinumab, Ixekizumab) respektive eine duale Hemmung von IL-17A und IL17F (Bimekizumab) bewirken (Kasten).
Neben den verschiedenen Biologika-Klassen (TNFα-, IL-17-, IL-12/23- oder IL-23- Antagonisten) und den konventionellen Systemtherapeutika (Ciclosporin A, Methotrexat, Acitretin oder Fumarsäureester) spielen auch niedermolekulare Wirkstoffe eine Rolle in der medikamentösen Therapie der Psoriasis. Apremilast ist ein oraler PDE-4(Phosphodiesterase-4)-Hemmer, der unter anderem für die Behandlung der Psoriasis zugelassen ist. Die antientzündlichen Effekte von Apremilast werden durch eine Erhöhung des intrazellulären cAMP-Spiegels herbeigeführt. Überdies wird mit Roflumilast aktuell ein topisch anwendbarer PDE-4-Hemmer in klinischen Studien geprüft [34]. Dies gilt auch für weitere Verteter der «Small Molecules» (Kasten).
Literatur:
- van de Kerkhof PC: From Empirical to Pathogenesis-Based Treatments for Psoriasis. J Invest Dermatol 2022; 142(7): 1778–1785.
- Di Meglio P, Villanova F, Nestle FO: Psoriasis. Cold Spring Harb Perspect Med 2014; 4(8): 1–30.
- Prinz JC: Human Leukocyte Antigen-Class I Alleles and the Autoreactive T Cell Response in Psoriasis Pathogenesis. Front Immunol 2018; 9: 954.
- Schäkel K, Schön M, Ghoreschi K: Pathogenese der Psoriasis vulgaris. Der Hautarzt 2016; 67(6): 422–431.
- Di Cesare A, Di Meglio P, Nestle FO: The IL-23/Th17 axis in the immunopathogenesis of psoriasis. Journal of Investigative Dermatology 2009; 129(6): 1339–1350.
- Lynde CW, et al.: Interleukin 17A: toward a new understanding of psoriasis pathogenesis. Journal of the American Academy of Dermatology 2014; 71(1): 141–150.
- Johansen C, et al.: Inverse regulation of the nuclear factor-κB binding to the p53 and interleukin-8 κB response elements in lesional psoriatic skin. Journal of investigative Dermatology 2005; 124(6): 1284–1292.
- Eyerich S, et al.: Mutual antagonism of T cells causing psoriasis and atopic eczema.N Engl J Med 2011; 365: 231–238.
- Schneider S, Li L, Zink A: Psoriasis – Differentialdiagnosen und Therapie. Akt Rheumatol 2022; 47: 324–332.
- Greb JE, et al.: Psoriasis. Nature Reviews Disease Primers 2016; 2: 16082.
- Lowes MA, et al.: Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci U S A 2005; 102: 19057–19062.
- Conrad C, et al.: Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 2007; 13: 836–842.
- Bovenschen HJ, Seyger MM, van de Kerkhof PC: Plaque psoriasis vs. atopic dermatitis and lichen planus: a comparison for lesional T-cell subsets, epidermal proliferation and differentiation. Br J Dermatol 2005; 153: 72–78.
- Clark RA: Resident memory T cells in human health and disease. Sci Transl Med. 2015 Jan 7; 7(269): 269rv1.
- Cheuk S, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014; 192: 3111–3120.
- Benezeder T, Wolf P: Resolution of plaque-type psoriasis: what is left behind (and reinitiates the disease). Semin Immunopathol 2019; 41: 633–644.
- Prinz JC: Melanocytes: Target Cells of an HLA-C*06:02-Restricted Autoimmune Response in Psoriasis. J Invest Dermatol 2017; 137(10): 2053–2058.
- Arakawa A, et al.: Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 2015; 212(13): 2203–2212.
- Hawkes JE, Chan TC, Krueger JG: Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 2017; 140(3): 645–653.
- Armstrong AW, et al.: Comparison of biologics and oral treatments for plaque psoriasis: a meta-analysis. JAMA Dermatol 2020; 156: 258–269.
- Papp K, et al.: Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018; 379: 1313–1321.
- Lebwohl MG, et al.: Trial of roflumilast cream for chronic plaque psoriasis. N Engl J Med. 2020; 383: 229–239.
- Robbins K, et al.: Phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of plaque psoriasis. J Am Acad Dermatol 2019; 80: 714–772.
- Mohd Noor AA, Azlan M, Mohd Redzwan N: Orchestrated Cytokines Mediated by Biologics in Psoriasis and Its Mechanisms of Action. Biomedicines. 2022 Feb 20;10(2): 498. www.mdpi.com/2227-9059/10/2/498, (letzter Abruf 15.03.3023)
- Arzneimittelinformation, www.swissmedicinfo.ch, (letzter Abruf 15.03.2023)
- Ellis CN, et al.: Cyclosporine improves psoriasis in a double-blind study. JAMA 1986; 256: 3110–3116
- Griffiths CE, et al.: Clearance of psoriasis with low dose cyclosporin. Br Med J (Clin Res Ed) 1986; 293: 731–732.
- Abrams JR, et al.: Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plaques, including the activation of keratinocytes, dendritic cells, and endothelial cells. J Exp Med 2000; 192: 681–694.
- Nicolas JF, et al.: CD4 antibody treatment of severe psoriasis. Lancet 1991; 338: 321
- Wrone-Smith T, Nickoloff BJ: Dermal injection of immunocytes induces psoriasis. J Clin Invest. 1996; 98: 1878–1887.
- Nickoloff BJ, Wrone-Smith T: Injection of pre-psoriatic skin with CD4+ T cells induces psoriasis. Am J Pathol 1999; 155: 145–158.
- Zaba LC, et al. : Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses [published correction in J Exp Med 2008;205:1941]. J Exp Med. 2007; 204: 3183–3194.
- Zaba LC, et al.: et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signalling, not immediate response TNF genes. J Allergy Clin Immunol. 2009; 124: 1022–1030.E395
- Lebwohl MG, et al.: Trial of roflumilast cream for chronic plaque psoriasis. N Engl J Med. 2020; 383: 229–239.
DERMATOLOGIE PRAXIS 2023; 33(2): 20–22
Autoren
- Mirjam Peter, M.Sc.
Publikation
- DERMATOLOGIE PRAXIS


Comments are closed.